How to use the Investigation Flowchart
Laboratory Analysis
Investigations of "Out of Specification (OOS) / Out of Trend (OOT)/ Atypical results" have to be done in cases of:
- Batch release testing and testing of starting materials.
- In Process Control testing: if data is used for batch calculations/decisions and if in a dossier and on Certificates of Analysis.
- Stability studies on marketed batches of finished products and or active pharmaceutical ingredients, on going / follow up stability (no stress tests)
- Previous released batch used as reference sample in an OOS investigation showing OOS or suspect results.
- Batches for clinical trials.
- All solutions and reagents should be retained until all data has been second person verified as being within the defined acceptance criteria.
- Pharmacopoeia have specific criteria for additional analyses of specific tests (i.e. dissolution level specification for S1, S2 & S3 testing; Uniformity of dosage units specification for testing of 20 additional units; Sterility Testing)
- However if the sample test criteria is usually the first level of testing and a sample has to be tested to the next level this should be investigated as it is not following the normal trend.
- The OOS process is not applicable for In-process testing while trying to achieve a manufacturing process end point i.e. adjustment of the manufacturing process. (e.g. pH, viscosity), and for studies conducted at variable parameters to check the impact of drift (e.g. process validation at variable parameters).
OOS / OOT Result
Out of Specification (OOS) Result
- Test result that does not comply with the pre-determined acceptance criteria (i.e. for example, filed applications, drug master files, approved marketing submissions, or official compendia or internal acceptance criteria).
- Test results that fall outside of established acceptance criteria which have been established in official compendia and/or by company documentation (i.e., Raw Material Specifications, In-Process/Final Product Testing, etc.).
Out of Trend (OOT) Result
- Is generally a stability result that does not follow the expected trend, either in comparison with other stability batches or with respect to previous results collected during a stability study. However the trends of starting materials and in process samples may also yield out of trend data.
- The result is not necessarily OOS but does not look like a typical data point.
- Should be considered for environmental trend analysis such as for viable and non viable data (action limit or warning limit trends).
- Results that are still within specification but are unexpected, questionable, irregular, deviant or abnormal. Examples would be chromatograms that show unexpected peaks, unexpected results for stability test point, etc.
Phase la Investigation
Definition
- Phase la investigation is to determine whether there has been a clear obvious errors due to external circumstances such as power failure or those that the analyst has detected prior to generating data such as spilling sample that will negate the requirement of a Phase Ib investigation.
- For microbiological analysis this may be after the analysis has been completed and reviewed during reading of the samples.
- It is expected that these issues are trended even if a laboratory investigation lb or ll was not raised.
Obvious Error
Examples
Calculation error: analyst and supervisor to review, both initial and date correction.
Power outage: analyst and supervisor document the event, annotate “power failure; analysis to be repeated” on all associated analytical documentation.
Equipment failure: analyst and supervisor document the event, annotate “equipment failure; analysis to be repeated” cross reference the maintenance record.
Testing errors: for example, spilling of the sample solution, incomplete transfer of a sample; the analyst must document immediately.
for microbiology it could be growth on a plate not in the test sample area, negative or positive controls failing.
Incorrect Instrument Parameters: for example setting the detector at the wrong wavelength, analyst and supervisor document the event, annotate “incorrect instrument parameter”; analysis to be repeated” on all associated analytical documentation .
If no error was noted, and none of the above conditions were met Phase Ib investigation must take place.
Phase Ib Investigation
Definitions
Specification
A specification is defined as a list of tests, references to analytical procedures, and appropriate acceptance criteria which are numerical limits, ranges, or other criteria for the tests described. It establishes the set of criteria to which a drug substance, drug product or materials at other stages of its manufacture should conform to be considered acceptable for its intended use. “Conformance to specification” means that the drug substance and drug product, when tested according to the listed analytical procedures, will meet the acceptance criteria. Specifications are critical quality standards that are proposed and justified by the manufacturer and approved by regulatory authorities as conditions of approval.
Regulatory Approved Specification
Specifications for release testing. If no release specifications have been established then the internal specification becomes the release specification.
Acceptance Criteria
Numerical limits, ranges, or other suitable measures for acceptance of the results of analytical procedures which the drug substance or drug product or materials at other stages of their manufacture should meet.
Internal Specification
Are also action limits within regulatory specifications.
Assignable Cause
An identified reason for obtaining an OOS or aberrant/anomalous result.
No Assignable Cause
When no reason could be identified.
Invalidated test
A test is considered invalid when the investigation has determined the assignable cause.
Reportable Result
Is the final analytical result. This result is appropriately defined in the written approved test method and derived from one full execution of that method, starting from the original sample.
Warning Level or Trend excursions
If two or more consecutive samples exceed warning (alert), or if an increasing level of counts, or same organisms identified, over a short period was identified consideration should be given to treat the results as action level excursions.
Hypothesis/Investigative Testing
Is testing performed to help confirm or discount a possible root cause i.e what might have happened that can be tested: for example it may include further testing regarding sample filtration, sonication /extraction; and potential equipment failures etc. Multiple hypothesis can be explored.
Investigation by Analyst and Supervisor
- Initial Investigation conducted by the analyst and supervisor using the Laboratory Investigation Checklist
- For microbiological analysis where possible once a suspect result has been identified ensure all items related to the test failure are retained such as other environmental plates, dilutions, ampoules/vials of product, temperature data, auto pipettes, reagents growth media. No implicated test environmental plates should be destroyed until the investigation has been completed.
- The Analyst and Supervisor investigation should be restricted to data / equipment / analysis review only: On completion of the Analyst and Supervisor investigation re measurement can start once the hypothesis plan is documented and is only to support the investigation testing.
- This initial hypothesis testing can include the original working stock solutions but should not include another preparation from the original sample (see: re testing).
ALSO READ: SOP for Handling Out of Specification (OOS)
Investigation by Analyst and Supervisor
The checklist may not be all inclusive, but should be a good guideline to cover the pertinent areas that need to be covered in any laboratory investigation:
- Correct test methodology followed e.g.. Version number.
- Correct sample(s) taken/tested (check labels was it taken from correct place).
- Sample Integrity maintained, correct container and chain of custody (was there an unusual event or problem).
- How were sample containers stored prior to use
- Correct sampling procedure followed e.g. version number
- Assessment of the possibility that the sample contamination has occurred during the testing/ re testing procedure ( e.g. sample left open to air or unattended).
- All equipment used in the testing is within calibration date.
- Review equipment log books.
- Appropriate standards used in the analysis.
- Standard(s) and/or control(s) performed as expected.
- System suitability conditions met (those before analysis and during analysis).
- Correct and clean glassware used.
- Correct pipette / volumetric flasks volumes used.
- Correct specification applied.
- Media/Reagents prepared according to procedure.
- Items were within expiry date
- A visual examination (solid and solution) reveals normal or abnormal appearance
- Data acceptance criteria met.
- The analyst is trained on the method.
- Interview analyst to assess knowledge of the correct procedure.
- Examination of the raw data, including chromatograms and spectra; any anomalous or suspect peaks or data.
- Any previous issues with this assay.
- Other potentially interfering testing/activities occurring at the time of the test.
- Any issues with environmental temperature/humidity within the area whilst the test was conducted.
- Review of other data for other batches performed within the same analysis set.
- Consideration of any other OOS results obtained on the batch of material under test.
- Assessment of method validation.
Additional considerations for microbiological analysis:
- Are the isolates located as expected on glove dab marks, SAS ‘dimples’, filter membrane etc.
- Was the sample media integral i.e. no cracks in plates.
- Was there contamination present in other tests (or related tests) performed at the same time, including environmental controls.
- Were negative and positive controls satisfactory.
- Were the correct media/reagents used.
- Were the samples integral (not leaking)
- Were the samples stored correctly (refrigerated)
- Were the samples held for the correct time before being tested.
- Was the media/reagent stored correctly before use
- Were the incubation conditions satisfactory.
- Take photographs to document the samples at time of reading (include plates, gram stains and any thing else that may be relevant).
ALSO READ: SOP for Handling of Out of Trend (OOT)
Phase II Investigation
- Phase II investigation Conducted when the phase I investigations did not reveal an assignable laboratory error. Phase II investigations are driven by written and approved instructions against hypothesis. Prior to further testing a manufacturing investigation should be started to determine whether there was a possible manufacturing root cause.
- If not already notified the contract giver/MAH/QP (in accordance with the responsibilities in the TA) should be notified along with production and QA if a manufacturing site.
- It is important when considering performing additional testing that it is performed using a predefined retesting plan to include retests performed by an analyst other than the one who performed the original test. A second analyst performing a retest should be at least as experienced and qualified in the method as the original analyst.
- If the investigation determines analyst error all analysis using the same technique performed by the concerned analyst should be reviewed.
Definitions
Hypothesis/Investigative Testing
Is testing performed to help confirm or discount a possible root cause i.e what might have happened that can be tested: for example it may include further testing regarding sample filtration, sonication /extraction; and potential equipment failures etc. Multiple hypothesis can be explored.
Re-Test
Performing the test over again using material from the original sample composite, if it has not been compromised and/or is still available. If not, a new sample will be used.
Re-sample
A new sample from the original container where possible, required in the event of insufficient material remaining from original sample composite or proven issue with original sample integrity.
Most probable cause
Scientifically justified determination that the result appears to be laboratory error.
Unknown Cause / No Assignable Cause
Hypothesis Testing (Applicable to Phase Ia and Phase II):
- Should be started as part of Phase Ia and continue into Phase II if no assignable cause found.
- Description of the testing should be written, and then approved by QA/Contract Giver/QA equivalent prior to initiating investigational testing. The requirements of investigational testing listed below:
- The description must fully document
- The hypothesis to the test the root cause being investigated.
- What samples will be tested.
- The exact execution of the testing.
- How the data will be evaluated
- This Hypothesis testing may continue from the re measurement of the original preparations.
- Investigational testing may not be used to replace an original suspect analytical results. It may only be used to confirm or discount a probable cause.
- If no assignable cause that could explain the results can be identified during the manufacturing investigation or the assay failure investigation retesting may be considered. Part of the investigation may involve retesting a portion of the original sample.
Retesting:
- Performed on the original sample not a different sample.
- Can be a 2nd aliquot from the same sample that was the source of the original failure.
- If insufficient quantity of the original sample remains to perform all further testing then the procedure for obtaining a resample must be discussed and agreed by QA/Contract Giver/QA equivalent. The process of obtaining the resample should be recorded within the laboratory investigation.
- The decision to retest should be based on sound scientific judgement. The test plan must be approved before re testing occurs.
- The minimum number of retests should be documented within the procedure and be based upon scientifically sound principles. Any statistical review with regards to %RSD and repeatability should relate to the values obtained during method validation (accuracy, precision, and intermediate precision). The number of retests should be statistically valid.
- The retests should be performed by a different analyst where possible. The second analyst should be at least as experienced and qualified in the method as the original analyst.
Averaging:
- The validity of averaging depends upon the sample and its purpose. Using averages can provide more accurate results. For example, in the case of microbiological assays, the use of averages because of the innate variability of the microbiological test system. The kinetic scan of individual wells, or endotoxin data from a number of consecutive measurements, or with HPLC consecutive replicate injections from the same preparation (the determination is considered one test and one result), however, unexpected variation in replicate determinations should trigger investigation and documentation requirements.
- Averaging cannot be used in cases when testing is intended to measure variability within the product, such as powder blend/mixture uniformity or dosage form content uniformity.
- Reliance on averaging has the disadvantage of hiding variability among individual test results.
- For this reason, all individual test results should normally be reported as separate values. Where averaging of separate tests is appropriately specified by the test method, a single averaged result can be reported as the final test result. In some cases, a statistical treatment of the variability of results is reported. For example, in a test for dosage form content uniformity, the standard deviation (or relative standard deviation) is reported with the individual unit dose test results.
- In the context of additional testing performed during an OOS investigation, averaging the result (s) of the original test that prompted the investigation and additional retest or resample results obtained during the OOS investigation is not appropriate because it hides variability among the individual results. Relying on averages of such data can be particularly misleading when some of the results are OOS and others are within specifications. It is critical that the laboratory provide all individual results for evaluation and consideration by Quality Assurance.
- All test results should conform to specifications (Note: a batch must be formulated with the intent to provide not less than 100 percent of the labelled or established amount of the active ingredient.
- Averaging must be specified by the test method.
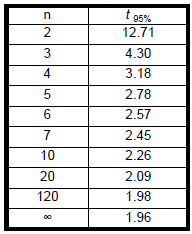
- Consideration of the 95% Confidence Limits (CL 95%) of the mean would show the variability when averaging is used.
- Consideration of using 95% Confidence Limits (CL 95%95%) of the mean would show the variability when averaging is used.
- The confidence interval is calculated from the formula:
CL= sample mean ± t 95% sample standard deviation / √n
Where,
t is a value obtained from tables
n is the sample size
Table:
Re-sampling:
- Should rarely occur!
- If insufficient quantity of the original sample remains to perform all further testing then the procedure for obtaining a resample must be discussed and agreed by QA/Contract Giver/QA equivalent. The process of obtaining the resample should be recorded within the laboratory investigation.
- Re sampling should be performed by the same qualified methods that were used for the initial sample. However, if the investigation determines that the initial sampling method was in error, a new accurate sampling method shall be developed, qualified and documented.
- It involves the collecting a new sample from the batch.
- Will occur when the original sample was not truly representative of the batch or there was a documented/traceable lab error in its preparation.
- Evidence indicates that the sample is compromised or invalid.
- Sound scientific justification must be employed if re sampling is to occur.
Outlier test:
- An outlier may result from a deviation from prescribed test methods, or it may be the result of variability in the sample. It should never be assumed that the reason for an outlier is error in the testing procedure, rather than inherent variability in the sample being tested.
- Statistical analysis for Outlier test results can be as part of the investigation and analysis. However for validated chemical tests with relatively small variance and that the sample was considered homogeneous it cannot be used to justify the rejection of data.
- While OOS guidance is not directly intended for bioassay analysis, it can be used as a starting point for the investigation. Compendia such as the BP; PhEur and USP, provide guidance on outliers for these types of analysis.
Microbiological Investigations:
- These are difficult to perform as the result can be 1 to 2 weeks after the analysis was performed and may be weeks after the batch was manufactured.
- It is important to evaluate the test conditions carefully and determine what the boundary of samples/products/manufacturing area is. It you do not determine the boundary of the suspect results it is difficult to determine if it one or more batches impacted.
- The laboratory and manufacturing investigations need to be in depth.
- The investigations should clearly state the hypothesis and who will be responsible for the identified tasks.
- Are the organisms of an expected type, determine likely source would it be likely to be found where it was?
- Review the media prepared in house or bought in pre prepared, supplier history, sterilization history.
- Equipment/utilities used validation, maintenance and cleaning status.
- Evaluate area/environmental trends for test area and support areas.
- Cleaning and maintenance of the test environment
- Disinfectant used
- Use appropriate root cause analysis to help brain storm all possibilities.
- It is likely that there may be more than one root cause.
- Review decisions and actions taken in light of any new information.
- Due to the variability of microbiological results don’t limit the investigation to the specific batch it should be broader to review historical results and trends.
- Unusual events should be included to understand potential impacts.
- What is the justification to perform a repeat analysis (is sample left); re test or resample.
- Any identifications may need to be at DNA/RNA level (bioburden failures).
- All potential sources of contamination need to be considered process flow the issue from sample storage to the test environment.
- Use scientific decisions/justifications and risk based analysis.
- The investigation may include working closely with the manufacturing team.
- During the investigation it is an advantage to go and look at where the contamination occurred.
- Ask how relevant plant is cleaned, tested for integrity, checked for wear, checked for material suitability and maintained at the occurrence site may reveal possible causes.
- Where possible talk directly to the staff involved as some information may be missed if not looked at from the chemist/ microbiologist point of view.
- Look for other documentation such as deviations and engineering notifications around the area of concern (this is applicable to the laboratory as well as manufacturing).
- Trending can have species drift which may also be worthy of an action limit style investigation.
- Statistical analysis for microbiology can include lots of zero results so recovery rates or similar may have to be used.
- If a sample is invalidated the remaining level of assurance needs to be carefully considered, is their sufficient residual information?
- Corrective actions may be appropriate for more than one root cause.
Stability-OOS/OOT
- Stability OOS/OOT situations should be escalated as soon as the suspect result is found. Follow the investigation as above for Phase I and Phase II. For OOS Situations Regulatory agencies will require notification within a short time point of discovery due to recall potential.
- If abnormal results are found at any stability interval which predicts that the test results may be OOS before the next testing interval, schedule additional testing before the next scheduled testing interval. This will help better determine appropriate actions to be taken.
- The stability OOS should link to the Product Recall procedures.
OOT
- To facilitate the prompt identification of potential issues, and to ensure data quality, it is advantageous to use objective (often statistical) methods that detect potential out of trend (OOT) stability data quickly.
- OOT alerts can be classified into three categories to help identify the appropriate depth for an investigation. OOT stability alerts can be referred to as:
- Analytical,
- Process control, and
- Compliance alerts,
- As the alert level increases from analytical to process control to compliance alert, the depth of investigation should increase.
Stability
- A compliance alert defines a case in which an OOT result suggests the potential or likelihood for OOS results to occur before the expiration date within the same stability study (or for other studies) on the same product.
- The stability OOS should link to the Product Recall procedures.
- Historical data are needed to identify OOT alerts.
- An analytical alert is observed when a single result is aberrant but within specification limits i.e., outside normal analytical or sampling variation and normal change over time).
Phase III Investigation
- If the batch is rejected there still needs to be an investigation.
- To determine:
- if other batches or products are affected.
- identification and implementation of corrective and preventative action.
- The phase 3 investigation should review the completed manufacturing investigation and combined laboratory investigation into the suspect analytical results, and/or method validation for possible causes into the results obtained.
- To conclude the investigation all of the results must be evaluated.
- The investigation report should contain a summary of the investigations performed; and a detailed conclusion.
- For microbiological investigations, where appropriate, use risk analysis tools to support the decisions taken and conclusions drawn. It may not have been possible to determine the actual root cause therefore a robust most probable root cause may have to be given.
- The batch quality must be determined and disposition decision taken.
- Once a batch has been rejected there is no limit to further testing to determine the cause of failure, so that corrective action can be taken.
- The decision to reject cannot be reversed as a result of further testing.
- The impact of OOS result on other batches, on going stability studies, validated processes and testing procedures should be determined by Quality Control and Quality Assurance and be documented in the conclusion, along with appropriate corrective and preventive actions.
Batch Disposition
Conclusion
- If no laboratory or calculation errors are identified in the Phase I and Phase II there is no scientific basis for invalidating initial OOS results in favour of passing retest results. All test results, both passing and suspect, should be reported (in all QC documents and any Certificates of Analysis) and all data has to be considered in batch release decisions.
- If the investigation determines that the initial sampling method was inherently inadequate, a new accurate sampling method must be developed, documented, and reviewed and approved by the Quality Assurance responsible for release. A consideration should be given to other lots sampled by the same method.
- An initial OOS result does not necessarily mean the subject batch fails and must be rejected. The OOS result should be investigated, and the findings of the investigation, including retest results, should be interpreted to evaluate the batch and reach a decision regarding release or rejection which should be fully documented
- In those cases where the investigation indicates an OOS result is caused by a factor affecting the batch quality (i.e., an OOS result is confirmed), the result should be used in evaluating the quality of the batch or lot. A confirmed OOS result indicates that the batch does not meet established standards or specifications and should result in the batch's rejection and proper disposition. Other lots should be reviewed to assess impact.
- For inconclusive investigations in cases where an investigation:
- does not reveal a cause for the OOS test result and
- does not confirm the OOS result
- The OOS result should be given full consideration (most probable cause determined) in the batch or lot disposition decision by the certifying QP and the potential for a batch specific variation also needs considering.
- Any decision to release a batch, in spite of an initial OOS result that has not been invalidated, should come only after a full investigation has shown that the OOS result does not reflect the quality of the batch. In making such a decision, Quality Assurance/QP should always err on the side of caution.
Tags
Quality Assurance